INTRODUCCION
En la actualidad, el método más utilizado en sistemática es el cladístico. La formulación original de este método suele atribuirse al entomólogo (Dipterologo) alemán Willi Hennig, pero muchas de las ideas básicas del método fueron desarrolladas independientemente por varios investigadores (algunos mucho antes que Hennig). Hennig desarrollo sus ideas entre los años 50's y 60's, y su metodología empezó a hacerse conocida en la década de los 70's. Sostenía que las clasificaciones deben estar basadas en la filogenia, y propuso un método que permitiera hacer hipótesis de filogenia a partir de las observaciones de las características actuales de los organismos. Fue uno de los científicos que más se mostraron en contra de la idea de que la sistemática era una especie de "arte"; la idea de que el sistemático simplemente mete las cosas en casilleros, sin necesidad ni posibilidad de justificar cómo son elejidos los casilleros.
Hennig insistió, en cambio, en que la sistemática debe verse como una ciencia, y que sus principios y fundamentos por lo tanto deben ser justificados y discutidos en una forma lógica y racional. El difundir este punto de vista fue probablemente una de las contribuciones más importantes de Hennig.
Hasta mediados de la década de los 70's, se consideró que el principio fundamental de la cladística era que las clasificaciones debían basarse exclusivamente en las relaciones filogenéticas (sin importar tanto cómo han llegado a descubrirse/deducirse esas relaciones). En esta época fue común que se presentara a la cladística como un "rompimiento epistemológico" con el pasado, representando un enfoque completamente nuevo de cómo hacer clasificaciones. Esto implicaba que los trabajos de sistemático "pre-hennigianos" podían ser ignorados, ya que trataban de problemas ahora considerados irrelevantes. Sólo al tomar las relaciones filogenéticas en lugar de tan sólo las similitudes y diferencias podía lograrse, supuestamente, una clasificación más "científica" en lugar de tan sólo "deductiva".
El mismo Hennig, en sus trabajos, parecía desdeñar a menudo el aspecto descriptivo de la sistemática. Sin embargo, se fue haciendo evidente a mediados de los 70's que el método cladístico puede verse tambien como una forma especial de "describir". Si el método fue originalmente diseñado con el propósito de reconstruir filogenias, su idea fundamental era que el estudio de la filogenia era una ciencia empírica y las conclusiones debían basarse en evidencia (caracteres). Por lo tanto, el método cladístico puede verse tambien como un estudio de caracteres, es decir, una forma de relacionar clasificaciones y caracteres... y por lo tanto, como una forma especial de usar las clasificaciones para describir los caracteres.
Este punto de vista fue sostenido por autores como James Farris, Gareth Nelson y Norman Platnick. Según estos, el modo en que los caracteres se relacionan con la clasificación final en el método cladístico es el mejor, en lo que se refiere a eficiencia descriptiva. Desde este punto de vista la cladística no representa ningun "rompimiento epistemológico" con el pasado, sino tan sólo una manera más explícita y lógica de llevar a cabo el mismo propósito que han buscado todos los sistemáticos desde Linneo: Organizar a la diversidad en un sistema coherente. Los trabajos de sistemáticos "pre-hennigianos" tambien pueden (y deben) ser considerados, analizados y discutidos (Goloboff 1998).
CARACTERES
El análisis cladístico consiste en tres procesos: (1) el descubrimiento y selección de carácteres y taxa, (2) la codificación de los carácteres, y (3) determinación de los cladogramas que mejor explican la distribución de los caracteres sobre los taxa.
Las operaciones de los análsis cladísticos son fuertemente influenciados por la selección y resolución de los taxa y caracteres. Los tipos de carácteres disponibles (esqueletos, partes blandas, ADN, etc) inevitablemente afectan la selección del método cladístico.
Actualmente, las matrices de datos están formados por carácteres con códigos discretos dispuestos en columnas y los taxa en las filas:

CLADOGRAMAS
DEFINICIONES
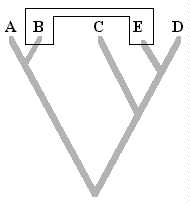
Un un árbol filogenético (cladograma), es una representación gráfica de las relaciones genealógicas entre taxa que están siendo determinadas por un investigador. En otras palabras, un árbol filogenético es una hipótesis de las relaciones genealógicas sobre un taxon. Veamos la siguiente figura:
 |
Se observan cuatro taxa: A...C, llamados Taxa terminales. Luego el taxon A es el Grupo Externo: Aquel taxon o grupo utilizado en el análisis que no esta incluido en el grupo de estudio. Se utiliza con propósitos comparativos. El grupo externo más importante es el Grupo Hermano, el cual puede llevar de muchos estudios filogenéticos para su determinación. Por lo general se utilizan varios grupos externos en los análisis cladísticos.
Seguidamente se aprecian líneas que conectan a los taxa terminales, a estas se les conoce como ramas. La union entre dos ramas forma un nodo, y representa un evento de especiación. Un internodo es una línea que conecta dos eventos de especiación y representa al menos a una especie ancestral (hipotética). Un internodo localizado en la parte más basal del cladograma se conoce como raíz.
Otra forma de representar las relaciones entre A, B, C y D es mediante un diagrama de Venn:
 |
Por último, la formas más sencillas para representar las relaciones entre los taxa A, B, C y D, es con teoría de conjuntos (A u B u C u D) o bien por notación parentética (A (B (CD))).
GRUPOS MONOFILETICOS, PARAFILETICOS Y POLIFILETICOS
Monofilético: Es un grupo que incluye a todos los ancestros y a sus descendientes. Los miembros de un grupo monofilético tienen un conjunto de relaciones de ancestría no compartidas con ninguna otra especie colocada fuera del grupo. En otras palabras, es una unidad histórico-evolutiva. (ver figura)


Parafilético: Es un grupo que se "origina" una sola vez pero excluye a uno o más de sus descendientes. (ver figura)


Polifilético: Es un grupo que se "origina" varias veces, o dicho de otra forma posee varios ancestros. (ver figura)

La diferencia fundamental entre grupos monofiléticos y los no-monofiléticos, es que los segundos forman clasificaciones no naturales o artificiales, es decir, no forman parte de un proceso evolutivo o con una historia evolutiva común. En algunos casos es díficil distinguir claramente el estatus de un grupo como parafilético o polifilético.
Un cladograma puede realizarse manualmente o mediante un programa de computación (software)
Además los caracteres de un cladograma deben ser optimizados
CLADOGRAMAS OBTENIDOS MANUALMENTE
Los cladogramas se construyen encontrando los grupos hermanos dentro de un grupo monofilético. Esto se hace por medio de la búsqueda de las sinapomorfías más inclusivas.
CLADOGRAMAS OBTENIDOS CON PROGRAMAS
Existen programas que permiten obtener los cladogramas por computación. Si bien los mismos no solucionan ningún problema que no se pudiera resolver por métodos manuales, resuelven cladogramas muy complejos y lo hacen rápidamente; esta rapidez permite que puedan, además, probarse distintas opciones de búsqueda y tratamiento de los caracteres. También brindan una lista de los cambios que sufren los caracteres a lo largo del cladograma y de las homplasias y apomorfías. Aunado a esto los cambios pueden graficarse en cada árbol y los cladogramas exportarse como imagenes a programas tan comunes para nosotros como MS-POWER POINT y MS-WORD.
La mayoría de los programas funcionan utilizando la técnica de los árboles de Wagner, la que utiliza el concepto de parsimonia para conseguir el árbol filogenético con menor número de cambios evolutivos. Esto se logra mediante una optimización de los caracteres, es decir, "mapeando" los caracteres sobre un cladograma para obtener el árbol que necesita el menor número de pasos.
OPTIMIZACIÓN GENERALIZADA
La optimizacion de cualquier tipo de carácter toma dos pasadas: la primera hacia abajo y la segunda hacia arriba. En la pasada hacia abajo se calcula para cada nodo un conjunto preliminar de estados. Estos conjuntos se encuentran en forma tal que produzcan longitud minima dados los descendientes del nodo. La pasada hacia abajo permite calcular la longitud del árbol; cada vez que no es posible asignar a un nodo un estado tal que no produzca nigun cambio a lo largo de las ramas descendientes, se incrementa la longitud del arbol en el número correspondiente de pasos. Este número de pasos es independiente del "peso" del caracter; si se le da al carácter mayor peso, el número de pasos debe ser multiplicado por el peso (cada paso del carácter "vale" por varios). Dado que la optimización se lleva a cabo sin tomar en cuenta el peso, el hecho de pesar caracteres no afecta los estados que se asignen a los nodos, ni en que ramas del árbol ocurrir n cambios. La pasada hacia abajo toma en cuenta solo la información "por arriba" del nodo; si los grupos hermanos del nodo en cuestión tienen otros estados, sin embargo, puede ser necesario cambiar las asignaciones. Como el nodo raiz del arbol no tiene grupos hermanos, los estados calculados en la primer pasada para ese nodo son finales. Se empieza entonces la pasada hacia arriba, asignando a cada nodo estados finales, tomando en cuenta los estados (finales) del ancestro del nodo y los estados (preliminares) de los descendientes). Para cada estado final del ancestro, aquellas asignaciones al nodo que produzcan la longitud mínima (ancestro-nodo + nodo-descendientes) ocurriran en al menos una reconstrucción de máxima parsimonia (Goloboff 1998). Como ejemplo veamos...
Los seis taxa A...F, poseen c/u los estados de carácter A(1), B(0), C(1), D(1), E(0) y F(0), como se puede ver en la figura:

Empecemos la pasada hacia abajo...primero con el nodo que compromete a los taxa A y B. Entre los estados "1" y "0", solo se puede escoger como asignación más parsimoniosa a ambos "01". Debido a que si se coloca cualquiera de los dos el número de pasos será el mismo. Esto es una ambiguedad:  | |
Sigamos con el nodo de (A, B) y el taxon C. Entre el nodo "01" y el taxon C con el estado "1", la asigación más adecauda es "1":  | |
Entre el nodo (A, B, C) y el taxon D, con el estado de carácter "1", la asignación más adecuada es, de nuevo "1":  | |
Luego entre el nodo (A, B, C, D) y el taxon E, con el estado de carácter "0", la mejor opción es una ambiguedad "01":  | |
Finalmente, entre el nodo (A, B, C, D, E) y el taxon F, con el estado "0", la asignación adecuada es "0": |
Tenemos entonces estados parciales de los nodos, veamos ahora como se realiza la pasada hacia arriba:

Se empieza desde el nodo sigiente a la raiz y se "ve" la información que está por arriba y abajo. ¿Qué estado de carácter se debe colocar para que se tenga el menor número de pasos? Con "1": Se registran dos cambios, de 0->1 y luego de 1->0. Con "0": Se registra un solo cambio, de 0->1. Conclusión: Debo preferir "0" sobre la otra asignación, porque es la más parsimoniosa. | |
 En el siguiente nodo si coloco "1" tengo un paso (de 0->1). En cambio asignando "0" son dos pasos (de 0->1 y luego 0->1). Me quedo con "1"...  | |
A continuación se presenta una caso interesante ¿que pasará con la ambiguedad?. Si coloco "1", se cuentan cero pasos. Asumiendo que la ambiguedad ha tomado solo el estado "1", porque si fuera "0", debemos contar un paso (de 1->0). Si coloco "0", y en la ambiguedad se asume el estado "0", se obtiene tres pasos (de 1->0 y luego 0->1). Por el contrario si en la ambiguedad se elije "1" debemos contar tres pasos de 1->0 y luego 0->1 * dos veces). Conclusión, debo asignar el estado "1"...  | |
Finalmente nos queda ver que se le asigna al nodo que tiene la ambiguedad. Con "1",... solo se cuenta un paso.Y con "0",... dos. Se le asigna el estado "1".  | |
Este es el resultado final de la optimización, un árbol con las asinaciones de sus nodos representadas por la mínima longitud o menor número de cambios. Solo nos queda calcular el largo del árbol: De 0->1 un paso, y luego de 1->0 un paso, total 2 pasos. |

Si esto le parece sencillo, imagine calcular la optimización para 50 taxa y 60 o más caracteres. Recuerde que aquí Ud. acaba de optimizar 1 solo caracter, que para nuestra suerte era binario (0 1).
Tranquilo no se asuste, para resolver estos y otros inconvenientes tenemos los programas de computación.
Tranquilo no se asuste, para resolver estos y otros inconvenientes tenemos los programas de computación.
Existen otros tipos de optimización:
Fitch
Este método de optimización se usa solamente para carácteres no aditivos ("no ordenados"), es decir, cuando se considera que una transformación entre dos estados culaesquiera requiere el mismo número de pasos. Ejemplo: Para un carácter binario, pasa de 0->1 implica lo mismo que de 1->0.
Farris
Este algoritmo se utiliza para caracteres aditivos ("ordenados lineales"). En este caso, el costo de transformarse de un estado a otro es su difrencia númerica. Por ejemplo: Un caracter "X" con los estados "0", "1", y "2". El costo de pasar de 0->2, es la suma de pasar de 0->1 y luego de 1->2.
Dollo
Se utiliza cuando en ciertos casos no se permite la reversión de algun estado, debido a que las transformaciones son consideradas poco problables o imposibles. La optimización de Dollo fue creada debido a necesidad de mostrar escenarios a priori en los cuales los estados apomórficos solo se adquieran una vez, y que los homoplásicos sean considerados como una pérdida secundaria. Por ejemplo, en estudios morfológicos estructuras complejas como el ojo de los vertebrados, pudo solo presentarse una vez. De igual forma en el campo molecular, estudios empíricos sugieren que en el analsis de restricción de corte de la endonucleasa, para ADN, muestra una marcada asimetría entre la baja probabilidad de ganar un nuevo lugar y la alta de perderlo.
Carmin-Sokal
Al igual que la optimización de Dollo, es muy poco usada. Asume la baja probabilidadad de que una adquisición pueda perderse. La evolución de carácteres es irreversible.
GLOSARIO DE TÉRMINOS
A
ACCTRAN
Transformación acelerada (o "accelerated transformation"), un procedimiento para resolver optimizaciones ambiguas donde los caracteres con transformaciones de avance ("forward") son colocadas hacia la raiz del cladograma. En ACCTRAN la cantidad de homoplasia es realizada en términos de reversiones hacia la condición plesiomórfica.
ambiguedad
El resultado de la optimización de un carácter sobre un cladograma donde el largo de la rama es cero y por lo tanto un colapsamiento.
analogía
Son carácteres que se presentan funcionalmente similares pero su desarrollo (ontogenia-origen) y su estructura morfológica es diferente.
apomorfía
Un carácter derivado o estado de carácter.
árbol filogenético
Es una representación gráfica e histórica del curso de la especiación. En el sistema filogenético esto es cierto, aunque esten representados arboles de taxa supraespecíficos, debido a que cada uno de estos taxa es hipotetizado de tener un origen en una simple especie ancestral.
atributos
Son las diferentes representaciones que el carácter posee. Por ejemplo: al carácter "color de ojos" se le puede atribuir numerosas condiciones como "color rojo", "color verde", etc. Es un sinónimo de estados de carácter (o "Character state")
autapomorfía
Un carácter derivado o estado de carácter (apomorfía) que esta restringido a un simple taxon terminal. Una autapomorfía en ciertos casos puede ser una sinapomorfía a un nivel menos sensitivo. Una forma de carácter no informativo.
B
bootstrap
un procedimiento estadístico para estimar la varianza paramétrica de los datos observados, por medio del promedio de las varianzas de pseudoréplicas (muestras iguales o mayores). Los datos originales son muestreados con reemplazamiento para producir pseudomatrices que luego serán analizadas con algun método de búsqueda (arboles de Wagner), y finalmente se realiza un consenso estricto para ver la proporción (porcentaje) de grupos monofiléticos.
C
carácter
Es una hipótesis de homología primaria en dos o más taxa terminales, basados en observaciones originales de los organismos. Es un atributo observable de un organismo usado para distinguirlo de los otros
carácter binario
Cuando un carácter esta representado por solo dos atributos o estados de cáracter. Por ejemplo: El carácter "seta maxilar número uno (1-Mx)" posee dos estados "presente" o "ausente".
carácter continuo
Un carácter en donde los valores potenciales son infinitamente cerrados, por ejemplo: Largo del ala.
carácter multiestado
Cuando un carácter esta representado por más de un par de atributos.
cladogénesis
Es la evolución siguiendo un patrón ramificado divergente. Esto significa la evolución o generación de pares de especies a partir de especies ancestrales únicas.
cladograma
Un diagrama de entidades (taxon) que ilustra una forma ramificada, la cual esta basada en conexiones históricas entre ellas como es evidenciada por el compartimiento de caracteres derivados o sinapomorfías. Es decir, son una representación filogenética o un dendrograma histórico.
cladograma mínimo
El árbol más parsimonioso. Usando los métodos tradicionales, un cladogramá mínimo tiene mínima longitud. Se conoce también como cladograma óptimo.
clasificación
Es la serie de palabras usadas para representar un arreglo particular de organismos de acuerdo a algún principio o relación que se supone exite entre ellos.
clasificación filogenética
Es el arreglo de los organismos basados en sus relaciones genealógicas (histórica-filogenética) hipotéticas.
codificación
La conversión de obsercaciones originales en un formato discreto alfanumérico utilizado en el análsis
cladístico.
codificación aditiva
Un método para representar carácteres multiestado ordenados como, series unidas de carácteres binarios.
consenso
Un método para combinar un grupo de información contenida en varios cladogramas de los mismos taxa, en una sola topología, el árbol de consenso.
criterio de optimalidad
La suma total de todos los requerimientos a ser aplicados en un carácter durante la optimización, en términos de: orden, polaridad y dirección.
D
E
especie
Como taxón esta definido como linages que están separados de otros linajes en el sentido de que ellos evolucionan independientemente. Las especies comprenden el nivel más superior de la organización taxonómica sobre los cuales la evolución puede trabajar.
evolución reticular
Comprende el origen o formación de nuevas especies a partir de la hibridización (unión) de dos especies ancestrales preexistentes. Este patrón es bastante raro en animales, pero muy común en los procesos evolutivos de las plantas.
F
fenograma
Diagrama ramificado que ilustra una relación entre entidades (taxa) estimada por una similaridad total como única evidencia para su construcción. Este último término ha sido muy criticado por los "cladístas" ya que el uso de la totalidad de carácteres en forma redundante hace que: 1) los nodos son definidos por una mezcla de caracteres de avance (apomórficos) y primitivos (plesiomórficos), 2) no hay discriminación entre similaridad sinapomórfica (proveniente del compartimiento de un ancestro común) y similaridad producto de paralelismos y convergencias, y finalmente, 3) el sistema producido es sumamente inestable cuando son añadidos nuevos datos.
G
H
homología
Dos carácteres son homólogos, si uno procede directamente del otro. En este caso, tenemos una novedad evolutiva y su preexistente homólogo. Este par de homólogos se denominan serie de transformación evolutiva. El carácter original o preexistente es llamado plesimórfico (primitivo) y la novedad evolutiva es denominada carácter apomórfico (derivado).
homoplasia
Son carácteres que muestran similaridades estructurales (y entonces ontogenéticas) pero que se suponen han sido originadas independientemente uno del otro, ya sea de dos caracteres preexistentes o de un único carácter preexistente en dos épocas diferentes o en dos especies diferentes. Una homoplasia existe si los dos taxa que muestran el carácter tienen un acestro común que no posee dicho carácter.
I
índice de consistencia
Una medida de la cantidad de homoplasia de un carácter relativa, a un cladograma.
índice de consistencia re-escalado
Es el producto del índice de consistencia y el de retención, de un carácter.
índice de retención
Una medida de la cantidad de similaridad en un carácter que puede ser interpretado como una sinapomorfía de un cladograma. Es el grado de "sinapomorfía aparente".
J
jackknife
un procedimiento estadístico para estimar la varianza paramétrica de los datos observados, por medio del promedio de las varianzas de pseudoréplicas (muestras más pequeñas). Los datos originales son muestreados sin reemplazamiento para producir pseudomatrices que luego serán analizadas con algun método de búsqueda (arboles de Wagner), y finalmente se realiza un consenso estricto para ver la proporción (porcentaje) de grupos monofiléticos.
K
L
largo
El mínimo número de cambios de carácter (pasos) requeridos en un cladograma. La suma total del ajuste ("fit") de todos los carácteres en un cladograma.
M
N
O
optimización
Un procedimiento para reconstruir la secuencia de estados de carácteres más parsimoniosos sobre un cladograma, minimizando el criterio de optimalidad.
P
parsimonia
Este principio ha sido propuesto para ser utilizado cuando dos o más hipótesis competitivas resultan de un análisis de datos. Este principio establece que deberá escogerse aquella hipotésis que explique la distribución de caracteres de la forma más simple o económica. En otras palabras, la hipotésis preferible es aquella que exhibe la mayor congruencia en el patrón de carácteres sinapomórficos.
paso
Una ganancia o pérdida de un carácter o una transformación de un carácter multiestado sobre un cladograma.
pesado sucesivo
Un método iterativo para el pesado de carácteres "a posteriori" de acuerdo con su consistencia cladística, la cual es medida usualmente por el índice de consistencia o el índice de consistencia re-escalado.
pesos implicados
Un procedimiento de pesado de carácteres de acuerdo con su ajuste ("fit") en un cladograma, a partir del número de pasos extra implicados. La función de ajuste, es generalmente concava, dando más peso a aquellos carácteres con menos homoplasia. También es un método "a posteriori".
polaridad
Se dice que un carácter o serie de transformación están polarizados cuando la dirección de cambio del carácter o evolución ha sido especificada, por lo tanto se determina las plesiomorfías y apomorfías de los carácteres o estados de carácteres.
pseudoréplica
Una matriz de datos artificial producida por permutación de, o re-muestreo de, observaciones reales.
o
Q
R
S
serie de transformación
Una serie de tres o más carácteres o estados de carácteres apomórficos, incrementándose.
sistemática
Aquella rama de las Ciencias Biológicas encargada de clasificar (dar un orden) a la naturaleza. En otras palabras, trata sobre la teoría y práctica relacionada con la captura del ordenamiento en la naturaleza que es el resultado de patrones ancestros filogenéticos y descendientes.
subtree pruning and regrafting (SPR)
Un método de permutación de ramas ("Branch-swapping") que corta los subcladogramas del árbol entero y luego los coloca en nuevas posiciones del árbol.
stepwise addition
Secuencia por la cual los taxa son agregados en la construcción inicial del cladograma.
T
taxón (pl. taxa)
Es el agrupamiento de organismos al cual se le ha dado un nombre propio, o podría darse un nombre propio, pero este nombre no es decidido por pura conveniencia.
taxón natural
Una especie o grupo de especies (taxón supraespecífico) que existe en la naturaleza como resultado de una historia única de descendencia con modificación (= evolución).
tree bisection and reconnection (TBR)
Un método de permutación de ramas ("Branch-swapping") que corta los subcladogramas del árbol entero y lo re-enraiza para colocarlo en nuevas posiciones de las áreas remanentes del árbol.
U
V
W
X
Y
Z
BIBLIOGRAFÍA (TEXTOS EN ESPAÑOL)
Contreras-Ramos A, Goyenechea I. 2007. La sistemática, base del conocimiento de la biodiversidad. Colección Ciencia al Día. UAEH, Pachuca. 158p
De Luna E, Mishler BD. 1996. El concepto de homología filogenética y la selección de caracteres taxonómicos. Boletín de la Sociedad Botánica de México. 59: 131-146.
De Luna E, Guerrero JA, Chew-Taracena T. 2005. Sistemática biológica: avances y direcciones en la teoría y los métodos de la reconstrucción filogenética. Hidrobiológica. 15(3): 351-370.
Feliner GN. 1999. Tres décadas de Cladismo. Boletin de la Sociedad Entomológica Aragonesa. 26: 85-93.
Goloboff P. 1998. Principios Básicos de Cladística. Ed. Sociedad Argentina de Botánica. Buenos Aires, Argentina.
Hennig W. 1966. Elementos de una sistemática filogenética. Ed. Universitaria de Buenos Aires. Buenos Aires, Argentina.
Morrone JJ, Llorente-Bousquets J. 2002. Cuatro mitos acerca de la Cladistica. Dugesiana. 9(1): 47-51.
Morrone JJ. 2005. Sistemática, Biogeografía, Evolución: los patrones de la biodiversidad en tiempo-espacio. Ed. Las Prensas de Ciencias. Facultad de Ciencias UNAM, Mexico.
Scrocchi G, Dominguez E. 1992. Introducción a las Escuelas de Sistemática y Biogeografía. No. 40. Opera Lilloana. Fundación Miguel Lillo, Tucumán, Argentina.
No entiendo Nadaaaa...? buuu un nivel mas facil para enterderlo
ResponderEliminarSi. Esto es algo que hice hace mucho tiempo a partir de clases que tome, varios libros y una guia de un curso en Venezuela. Es mucha informacion... pero lo bueno es que existe mucha bibliografia en español e ingles. Te recomiendo el libro de Pablo Goloboff (el escribe super sencillo, y te aclara muchos conceptos basicos)...
EliminarHola Jonathan,
ResponderEliminarMi nombre es Jose Manuel
Yo entiendo algo, aunque no todo! Pero quiero ser un usuario de algun programa aunque no entienda las profundidades de como fue hecho dicho programa. Si, como es logico, debe entender lo basico de esta tecnica!!! Tengo en estudio un genero de diptera con 130 especies y estoy preparando la matris de caracteres que ya suman cerce de 80. He bajado el programa PHYLIP, pero no se como empezar, ni como cargar el programa ya que no veo en ninguna parte el menu que hablan en la descripcion de los programas. Podrias ayudarme dandome algunas luces. Te lo agradezco sobre manera!!!
Estimado JM Ayala, Gracias por tu comentario. Si quieres mándame un correo (jonathan.liria@gmail.com) y conversamos mejor. Yo puedo mandarte algunas cosas para que leas sobre cladismo.. y ejercicios para correr los datos....
EliminarMuy buena información. Gracias!
ResponderEliminar